Two molecular signaling pathways underlie the cardiac defects associated with one type of the inherited disorder Noonan syndrome (NS), researchers from Masonic Medical Research Institute, Beth Israel Deaconess Medical Center and Weill Cornell Medicine find in a new study. The investigators built the first patient-derived stem cell model of the RAF1 mutant form of NS to investigate the defect, a severe thickening of the heart muscle called hypertrophic cardiomyopathy, in these patients.
Noonan syndrome is a disorder characterized by unusual facial characteristics, short stature and a multitude of heart defects and caused by mutations in any one of several genes, each of which affects the function of a signaling pathway called the RAS-Mitogen Activated Protein Kinase (MAPK) pathway. This pathway is critical for the regulation of cell growth, differentiation and cell death. Patients with the RAF1 mutation usually develop a severe form of hypertrophic cardiomyopathy that is often lethal in infancy or childhood.
“NS is the most common of a group of rare congenital disorders termed RASopathies, which include Costello Syndrome, NS with multiple lentigines (formerly LEOPARD) Syndrome, Neurofibromatosis, Cardiofaciocutaneous Syndrome, and others, and has a prevalence of about 1:1,000 to 1:2,500 children born each year worldwide,” said senior author Dr. Maria Kontaridis, director of research at Masonic Medical Research Institute and associate professor of medicine at Beth Israel Deaconess Medical Center Harvard Medical School. “NS-associated RAF1 mutations specifically are even rarer, but the effects of this mutation are also much more severe than the other mutations thought to be causal to NS.”
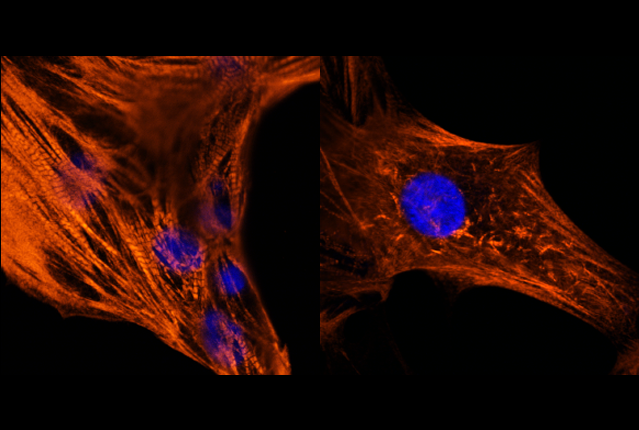
In the study, published June 5 in Circulation, the investigators reproduced the hallmark features of the heart cell defect in the lab using state-of-the art techniques to transform skin cells from a patient with Noonan syndrome with a RAF1 mutation into beating heart cells called cardiomyocytes. By examining these patient-derived cardiomyocytes, the investigators determined that aberrations in two different signaling pathways together cause the cells to become enlarged and exhibit other structural defects.
"Currently there is no treatment to prevent the onset of NS and protect against the heart defects that are associated with the RAF1 mutations that cause this disease,” said first author Dr. Fabrice Jaffré, instructor of cell and developmental biology in surgery at Weill Cornell Medicine. “Our studies sought to model and understand the molecular mechanisms that trigger the disease and its cardiac hallmarks so that we can identify new targeted treatments.”
The team began by converting skin cells from the Noonan syndrome patient into cells that can differentiate into any cell type in the body, called induced pluripotent stem cells (iPSCs). They were then able to instruct the iPSCs to develop into cardiomyocytes. These cells exhibited many of the features seen in the disease, including enlarged size and abnormal contractions.
Inside the cells, the signals controlling cell growth and function were also abnormal. The investigators determined that two pathways working in parallel led to the disease traits seen in RAF1 mutation patients. The first, named the ERK5 signaling pathway after one of the key proteins involved, was hyperactive, leading the cells to grow abnormally large. This was an unexpected finding because RAF1 wasn’t known to affect this pathway. In addition, the MEK1/2 signaling pathway was overly active, causing the structural defects that lead to contractile anomalies in these cells.
The results indicate that a combination therapy targeting both signaling pathways may be a promising way to treat the severe hypertrophic cardiomyopathy in patients with this type of Noonan syndrome. Currently, MEK1/2 inhibitors are used to treat some types of cancer, and additional drugs that block the ERK5 pathway are under development.
“Together, these data indicate that this disease is not linear; there are multiple, parallel signaling pathways that play important roles in leading to a disease phenotype. One pathway may be necessary, but alone it may not be sufficient to cause that disorder; therefore, use of a potential combinatorial therapy for each of these pathways identified herein could lead to a promising approach in treating NS-associated RAF1 patients with HCM,” Dr. Kontaridis said.
“Rare disease has always been a window to understanding more common disorders,” she added. “By studying rare diseases and delineating what mechanisms cause those diseases, we may reveal potential therapeutic options for other, more common, diseases as well.”
“We used to only rely on mouse studies to test new therapies for patients,” Dr. Jaffré said. “Now we can recreate their disease in a dish and determine how their own cells respond to treatment.”