A protein called Them1 prevents fat burning in cells by blocking access to the fuel source, according to preclinical research by Weill Cornell Medicine, NewYork-Presbyterian and Harvard Medical School/Beth Israel Deaconess Medical Center investigators. The findings may contribute to the development of a new type of obesity treatment that targets this response.
When cold temperatures force mice to ramp up fat burning to generate heat, they produce an enzyme that sits on standby, ready to shut down energy-intensive heat production when the need has passed, according a study published June 9 in Nature Communications. The study was a collaboration between Dr. David Cohen, chief of the Division of Gastroenterology and Hepatology at Weill Cornell Medicine and NewYork-Presbyterian/Weill Cornell Medical Center and the Vincent Astor Distinguished Professor of Medicine at Weill Cornell Medicine, and cell biology and microscopy expert Dr. Susan Hagen, associate professor of surgery at Harvard Medical School.
“The study explains a new mechanism that regulates metabolism,” said Dr. Cohen. “Them1 hacks the energy pipeline and cuts off the fuel supply to the energy-burning mitochondria.”
Dr. Cohen and his colleagues first became interested in Them1 about a decade ago when they discovered cold mice produce a lot of the enzyme in their brown fat tissue. They knew mice were using brown fat tissue to generate heat, so they assumed that Them1 was helping to produce heat. To prove this, they genetically engineered mice lacking Them1 to see what would happen.
“To our complete surprise, when you delete the gene for Them1, the mouse produces more heat, not less,” he said. In fact, the mice lacking the gene for Them1 burned so much energy trying to make heat that they ate twice as much as a typical mouse and still lost weight.
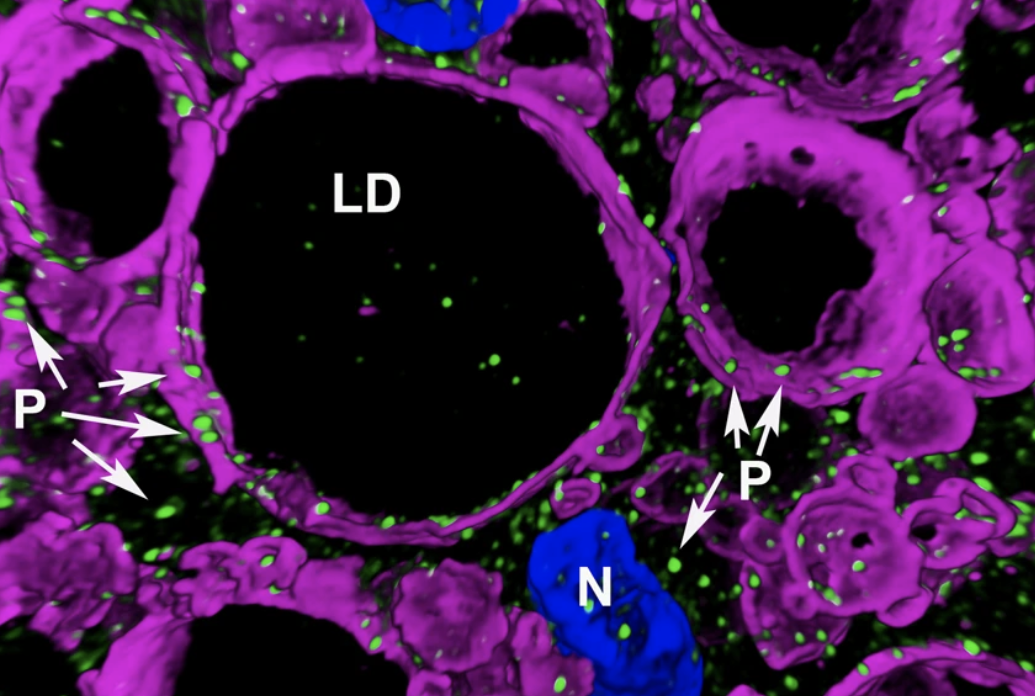
The new study helps explain how Them1 turns off heat production. In the experiments, the team used light and electron microscopy to observe Them1 in action in mouse brown fat cells grown in the laboratory. The experiments showed that when the cells are stimulated to burn fat using a drug, a phosphate group is added to Them1. The chemical modification causes Them1 molecules to spread out throughout the cell. This frees cellular powerhouses called mitochondria to efficiently turn the cell’s fat stores into energy. But when the stimulation stops, the Them1 molecules quickly reorganize like a protein “flash mob” into a membrane-less structure situated between the mitochondria and the fats they use as fuel, which stops energy production.
Humans also have brown fat and produce more Them1 in cold conditions, so the findings may have exciting implications for the treatment of obesity. Dr. Cohen and his colleagues are already working to develop an anti-obesity drug that inhibits Them1.
“If we could give humans a drug that inactivates Them1’s energy expenditure-suppressing activity, we might be able to increase fat burning,” Dr. Cohen said.